Adicionalmente, se presenta la sindáctila de los dedos 2, 3 y 4, los cuales pueden aparecer soldados al pulgar y al quinto dedo y dar la apariencia de manopla o manos en mitón, defectos que pueden observarse en los pies y que reducen la flexibilidad de estos. La unión de los dedos puede estar confinada solamente a los tejidos blandos o involucrar también la parte ósea. Las extremidades superiores se encuentran acortadas con aplasia o anquilosis de algunas articulaciones, especialmente de hombros, codos y cadera. (3, 6)
La afectación de las manos clínicamente se clasifica según su severidad en tres tipos (7): el tipo 1 presenta sindactilia del 2º- 3º y 4º dedos con el 1º y 5º separados; en el tipo 2 la fusión afecta al 2º-3º-4º y 5º dedos con el 1º separado, dando a la palma de la mano una forma de cuchara; en el tipo 3 la sindactilia es completa, afectando a los 5 dedos. El tipo más frecuente de mano en el Síndrome de Apert es el tipo 1. La clasificación de la sindactilia de los pies es similar a los tipos observados en las manos, aunque, al contrario de lo que ocurre en las manos, las uñas de los pies están al menos parcialmente segmentadas y la fusión de las falanges distales que se observa en las manos no ocurre en los pies.
En la piel se presenta hiperhidrosis generalizada y acné vulgaris en cara, pecho, espalda y miembros superiores; también se observa un exceso de arrugas en la piel que cubre la frente la cual es debido a la hipoplasia ósea del hueso frontal. Existen algunas afecciones relacionadas con el Síndrome de Apert como lo son defectos cardiovasculares, atresia pulmonar, ducto arterial permanente, fistula traqueoesofágica, estenosis pilórica, riñones poliquísticos, infecciones otológicas y apnea del sueño. Se encuentra afectada la flexibilidad de la columna cervical, debido a la fusión presente en las vertebras cervicales C5- C6. (3)
Es necesario establecer diagnóstico diferencial del Síndrome de Apert con otros síndromes que presentan craneosinostosis (Tabla 1) (1).
Desde el punto de vista genético, es aceptado que el origen del Síndrome de Apert es una mutación puntual esporádica en la gran mayoría de los casos, que produce una ganancia en la función del receptor 2 del factor de crecimiento de los fibroblastos. Esta mutación se encuentra en el cromosoma l0q26 y consiste en la sustitución en dos codones adyacentes, en las posiciones 755TCG que codifica para serina y 758CCT que codifica para prolina en el FGFR2; de modo que se sustituye una C por una G y se expresa de la siguiente manera: TCG→TGG y CCT→CGT, codificando las siguientes proteínas: Ser252Trp y Pro253Arg. (8, 9)
En cuanto al diagnóstico prenatal del Síndrome de Apert, la ecografía tridimensional introducida en la década de 1980 suministra una ayuda para examinar el útero materno en ángulos diferentes, lo que permite conocer los defectos estructurales del cráneo, cara y extremidades del feto. En centros de alta tecnología de los países desarrollados se pueden observar, mediante un ultrasonido a partir de la semana 20 de gestación, defectos craneales. (10)
El tratamiento que se encuentra disponible es sobre todo quirúrgico-paliativo, se basa principalmente en separar las sinostosis presentes en el cráneo y las sindactilias en los miembros pélvicos y torácicos.
Existen líneas de investigación sobre opciones de tratamiento médico. Una de estas es la proteína Noggin; dicha proteína antagoniza a la proteína ósea morfogenética tipo 4 (BMP4 por sus siglas en inglés). La proteína Noggin previene el cierre de las suturas craneales y su regulación se encuentra a la baja en el Síndrome de Apert. También se tiene la línea del calphostin C, que es un inhibidor de la proteincinasa C, elemento crucial en la señalización anómala en el Síndrome de Apert. El calphostin actúa mediante la modificación covalente del dominio regulatorio de la unión lipídica de la proteincinasa C, inhibiendo el cierre prematuro en las suturas craneales en modelos animales. (9)
Figura 1. Recién nacido con características fenotípicas de síndrome de Apert; nótese la craneosinostosis y la sindactilia en manos.
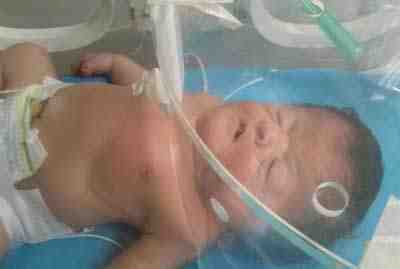
Fuente: propia del autor.
Figura 2. Craneosinostosis y cuello corto en recién nacido con características fenotípicas del síndrome de Apert.

Fuente: propia del autor.
Figura 3. Sindactilia en pies del recién nacido con características fenotípicas del síndrome de Apert.
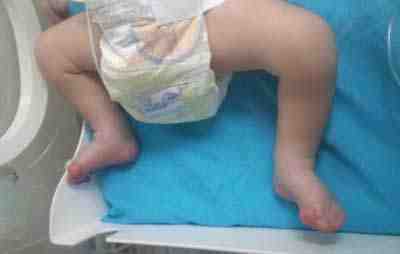
Fuente: propia del autor.
Tabla 1: Diagnósticos diferenciales del Síndrome de Apert.
Síndrome: Apert
Gen y Herencia: FGFR2
Locus 10q25-q26
AD
Características craneofaciales: Turribraquicefalia, hipoplasia del tercio medio de moderada a severa.
Características extremidades: Sindactilia simétrica (manos y pies), acortamiento rizomélico, anquilosis de codos. No hay polidactilia.
Síndrome: Crouzon
Gen y Herencia: FGFR2 (diferentes partes excepto exón B).
Locus: 10q25-q26
AD
Características craneofaciales: Craneosinostosis coronal, exoftalmos, hipertelorismo, hipoplasia del tercio medio facial, nariz en “pico”, orejas de implantación baja.
Características extremidades: Ninguna alteración.
Síndrome: Carpenter
Gen y Herencia: RAB23
Locus: 6p12.1-q12
AD
Características craneofaciales: Craneosinostosis, atrofia óptica, fisuras palpebrales antimongoloides, orejas grandes y de implantación baja.
Características extremidades: Anomalías de los dedos de las manos y pies: braquidactilia, polidactilia, sindactilia, clinodactilia.
Síndrome: Saether- Chotzen
Gen y Herencia: TWIST1 (se han registrado algunos con mutación en FGFR2).
Locus: 7p
AD
Características craneofaciales: Braquicefalia, sinostosis coronal bilateral asimétrica, hipertelorismo, ptosis palpebral, menos frecuentemente hendidura palatina.
Características extremidades: Anomalías de los dedos de las manos y pies: braquidactilia y sindactilia parcial.
Síndrome: Pfeifer Tipo II
Gen y Herencia: FGFR1
Locus: 8p11.2-p11.1
FGFR2
Locus: 10q26
AD
Características craneofaciales: Craneosinostosis en cráneo en forma de trébol asimetría craneofacial, hipoplasia maxilar, hipertelorismo, ptosis palpebral, estrabismo, paladar ojival.
Características extremidades: Pulgares fusionados y anchos, braquidactilia, anquilosis de codos y rodillas, polidactilia.