Presentación de un caso clínico. Síndrome de Gilbert
RESUMEN:
Analizaremos el caso de una persona de 40 años, de raza blanca, y que presenta alternativamente síntomas de fatiga y coloración amarillenta de la piel, varias veces, ictericia que aparece siempre, durante momentos de esfuerzo, estrés e infección. El síndrome de Gilbert se origina en una disminución de la disminución de excretar bilirrubina, por la célula hepática. Observaremos éste caso y veremos su concepto, su etiología, clínica, tratamiento y pronóstico.
Presentación de un caso clínico. Síndrome de Gilbert
Autor: Jiménez Cuadra, E. (Centro Salud Antequera), Martín José González Torres (Centro Salud Estación).
PALABRAS CLAVE: síndrome de Gilbert, paciente, medicina.
INTRODUCCIÓN: ANAMNESIS:
Imagen n. 1: Síndrome de Gilbert
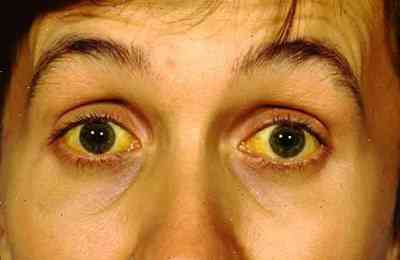
SÍNDROME DE GILBERT
Paciente de 40 años, que acude a consulta por síntomas alternativos, de ictericia, astenia, fatiga, a lo largo de los últimos años, y coloración verdosa de la piel, incluyendo las escleróticas, o sea la parte blanca del ojo. (1) Ya que en ésta enfermedad, algunas personas, tienden a presentar. en forma intermitente, coloración amarillenta, de las escleróticas, o de la piel, si tener otros síntomas de hígado. Esta enfermedad, tiende a llamarse, enfermedad o síndrome de Gilbert, afección benigna y frecuente del hígado.
Se observó (2) que la ictericia, aparecía y aumentaba en el sujeto, durante momentos de esfuerzo, estrés, infección y cuando no comen.
En resumen, (3) el elemento diagnóstico principal, es la hiperbilirrubinemia.
El diagnóstico diferencial, en éstos enfermos, y en éste paciente se hizo por un análisis de sangre, a partir de un aumento aislado de la bilirrubina indirecta, y la normalidad de las pruebas bioquímicas hepáticas.
PRUEBAS Y EXÁMENES REALIZADOS: Se realizó al paciente un perfil bioquímico de sangre y de hígado, y de bilirrubina, y se observaron cambios, que ocurren con ésta enfermedad.
El nivel de bilirrubina total, estaba levemente elevada, en su mayor parte era bilirrubina no conjugada. La bilirrubina conjugada, estaba en límites normales. Se (4) hizo una historia clínica detallada, incluyendo antecedentes personales de ictericia, y en el genograma, familia con historia de ictericia y enfermedad de Gilbert.
Examen físico, se hizo, haciendo un examen profundo del hígado y sistema gastrointestinal. La ictericia, puede ser detectada, clínicamente en los ojos, piel y mucosas orales.
La bilirrubina total, levemente elevada. Es bilirrubina no conjugada, en su mayor parte. La bilirrubina conjugada, estaba en límites normales.
Se le hizo un recuento un recuento sanguinario prescrito. A veces, esto puede indicar enfermedades infecciosas del hígado. Se encontró el conteo de reticulocitos, normal, siendo así enla enfermedad de Gilbert.
JUICIO DIAGNÓSTICO: Por la clínica, examen físico, recuentos sanguíneos y pruebas de función hepática, se diagnosticó este paciente, con un Síndrome de Gilbert.
PRONÓSTICO: La ictericia, va apareciendo y desapareciendo, a lo largo de su vida, no causa problemas de salud, pero puede confundir los resultados, de los análisis de la ictericia.
TRATAMIENTO: No se puso, porque no se necesita ningún tratamiento, para la enfermedad de Gilbert. El enfermo permanece estable, haciéndole un seguimiento, en Medicina de Familia y por el Digestivo.
DISCUSIÓN: No es una enfermedad rara. El Síndrome de Gilbert, es un trastorno hereditario hepático, caracterizado por una ictericia, debido (5) a un déficit parcial de la actividad enzimática, de la bilirrubina glucoronosiltrasferasa hepática, lo que provoca una hiperbilirrubinemia.
Revisaremos, diversos aspectos de ella.
CONCEPTO: Es un trastorno común hereditario, que afecta como la bilirrubina es procesada, en el hígado, y provoca que aparezca ictericia en la piel
PREVALENCIA: Entre el 3-10% de la población, está afectada. La ictericia neonatal, en recién nacidos, con el Síndrome de Gilbert, puede ser más grave y duradera, está ligada, entre un 20 y 30%, a una disminución de la actividad enzimática de la bilirrubina glucoronosiltransferasa. En las poblaciones, europea, americana y africana, se ha identificado una mutación del promotor del gen UGT1A1, familia de UDP, glucoronosiltransferasa, polipéptido A-1;2q37. Los pacientes que presentan la mutación, son homocigotos.
ETIOLOGÍA: Es hereditaria, un trastorno hereditario hepático, caracterizado por una ictericia, debida a un déficit parcial de la glucoronosiltransferasa.
Uno de los responsables de ésta afección, es el gen, UGT1.
Se ha demostrado que éste gen (6), tiene una pequeña