Caso clínico. Enfermedad de Canavan
RESUMEN:
La enfermedad de Canavan (EC) es un trastorno neurodegenerativo cuyo espectro varía entre formas graves con leucodistrofia, macrocefalia y retraso grave en el desarrollo, y una forma leve/juvenil muy poco frecuente caracterizada por un retraso leve del desarrollo.
Caso clínico. Enfermedad de Canavan
Autor: Jiménez Cuadra, Enriqueta (Centro Salud Antequera)
Palabras clave: enfermedad de Canavan, paciente, medicina.
Analizaremos un caso clínico, heredado de padre a hijo, cuyos síntomas aparecieron en el primer año de vida, como la falta de control en la cabeza, problemas en la alimentación.
INTRODUCCIÓN:
CASO CLÍNICO:
Presentamos un caso clínico, heredado de padre a hijo, cuyos síntomas aparecieron en el primer año de vida, como la falta de control en la cabeza, problemas en la alimentación, postura anormal con os brazos flexionados y las piernas estiradas, macrocefalia, irritabilidad, disminución del tono muscular, problemas que han ido aumentando a medida que el niño ha ido creciendo, vernos las pruebas y exámenes que se le hicieron, pronostico de enfermedad y tratamiento.
Etiología: En este caso la enfermedad de Canavan es hereditaria (1).
Se transmite de padres a hijos, y es más frecuente entre la población de judíos asquenacíes que en la población general.
Clínica: La leucodistrofia (degeneración de la substancia blanca cerebral) de Canavan es una enfermedad neurológica hereditaria rara, más frecuente en familias de origen judío.
Las leucodistrofias son un grupo de enfermedades desmielinizantes que presentan afectación primaria y predominante de la mielina (vaina de sustancia blanca que recubre los nervios) del sistema nervioso central (sistema formado por el encéfalo y la médula espinal), aunque en alguna de ellas se afecta además el sistema nervioso periférico (conjunto de nervios motores y sensitivos y ganglios situados fuera del encéfalo y la médula espinal). Se deben a un déficit enzimático (enzima es la sustancia proteica capaz de activar una reacción química del organismo) y tienen una base genética y hereditaria. Leucodistrofia, se requiere previamente excluir otras desmielinizaciones
a.- debidas a distrofia muscular congénita
b.- secundarias a procesos de otra naturaleza
c.- que ocurren en otros procesos, pero no forman parte de las manifestaciones clínicas predominantes, lo que sucede en enfermedades mitocondriales, como las enfermedades de Leigh y Leber (2)
d.- que afectan exclusiva o predominantemente al sistema nervioso periférico tales como neuropatías sensitivo-motoras hereditarias, polirradiculoneuritis desmielinizantes autoinmunes, etc.
Las leucodistrofias forman un grupo heterogéneo atendiendo a su origen: la enfermedad de Krabbe y la leucodistrofia metacromática son esfingolipidosis; la adrenoleucodistrofia es una enfermedad peroxisomal; la enfermedad de Pelizaeus Merzbacher se debe a un déficit de una proteína integrante de la mielina. La enfermedad de Canavan al efecto tóxico de una sustancia similar a un neurotransmisor (sustancia liberada por las terminaciones nerviosas bajo el influjo de una excitación, transmitiendo la información de una neurona a otra) de la corteza cerebral y la enfermedad de Alexander, a una anomalía del astrocito (célula neurológica en forma de estrella), ésta última se incluye tradicionalmente entre las leucodistrofias aunque la desmielinización sea secundaria y probablemente no sea hereditaria.
Se manifiestan fundamentalmente por alteraciones motoras y visuales. Las crisis convulsivas son raras y el retraso mental es de aparición tardía, apareciendo con la afectación axonal (el axón es la parte de la célula nerviosa que conduce impulsos procedentes del cuerpo celular de la neurona) secundaria. Los signos clínicos son comunes a todas ellas, aunque con ciertos rasgos diferenciales y están más en relación con la edad a la que se presenta la desmielinización que con la naturaleza de la misma.
En el lactante predomina la detención y retraso del desarrollo psicomotor (retraso en la adquisición de las habilidades que requieren la coordinación de la actividad muscular y mental), con irritabilidad, dificultad de alimentación y síndrome piramidal (parálisis de un lado del cuerpo, aumento de reflejos tendinosos y falta de reflejos cutáneos) que hace que el niño adopte una postura en opistótonos (espasmo tetánico de los músculos de la nuca y el dorso, que arquea el cuerpo que se apoya sólo en la nuca y los talones). Es frecuente la aparición de ceguera por atrofia (disminución de volumen y peso de un órgano) óptica.
A partir del primer año de vida, el síntoma inicial principal es la alteración de la marcha, que es atáxica (carencia de la coordinación de movimientos musculares) o espástica (contracción involuntaria y persistente de un músculo o grupo muscular) con hipotonía (tono anormalmente disminuido del músculo) axial, puede ser la única manifestación durante varios meses, hasta que poco a poco van apareciendo alteraciones de la conducta y del aprendizaje, como manifestaciones de deterioro cerebral.
A partir de los cinco años lo primero que aparecen son los síntomas mentales: problemas de comportamiento e hiperquinesia (actividad muscular exagerada) en la primera fase, seguidos de déficits de atención, concentración, aprendizaje y lenguaje. En una etapa posterior se desarrollan parálisis espásticas progresivas, movimientos anormales y espasmos tónicos, con evolución a un estado demencial (disminución irreversible de la facultad mental) y una rigidez de descerebración (estado que se produce cuando se extirpa o deja de funcionar el cerebro), que conduce a la muerte inexorablemente.(3)
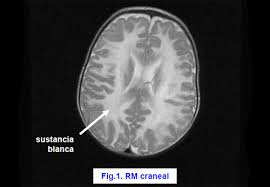
Enfermedad Canavan. Resonancia magnética.
– Reflujo de material alimenticio dentro de la nariz (regurgitación nasal)
– Problemas en la