
Los hamartomas retinianos o facomas, de color blanco nacarado, se observan situados a cierta distancia de la papila, estando compuestos por células gliales, fibroblastos y células ganglionares. Por estar colocados periféricamente en la retina, no causan trastornos visuales.
El hamartoma astrocítico del nervio óptico. Tiene escasa tendencia a crecer aunque sí se calcifica formando drusas. El sangrado de los vasos anómalos del tumor determina hemorragia vítrea. La angiografía fluoresceínica permite observar en el tiempo arterial el llenado rápido de los vasos dilatados intratumorales, y en la fase venosa la existencia de capilares tortuosos y dilatados.
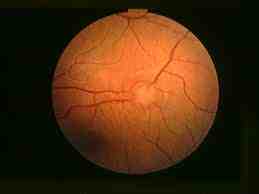

Hamartoma astrocítico
OTRAS MANIFESTACIONES
A nivel renal, la esclerosis tuberosa puede presentar quistes renales bilaterales y con una frecuencia de entre 50 a 70 % se presentan angiomiolipomas, tumor benigno de rara observación en la población general, si bien casi el 50% de los casos aparecen en pacientes con esclerosis tuberosa; constituidos por músculo liso, tejido adiposo y elementos vasculares.
Rabdomiomas benignos en el corazón, linfoangioleiomiomatosis en pulmón (frecuencia 1%), quistes en hígado, páncreas, ovarios, y otras vísceras son otras de las manifestaciones de esta enfermedad. En el esqueleto, se ha descrito una serie de alteraciones radiológicas clínicamente latentes: nódulos densos de contornos difusos en el cráneo y en las vértebras, islotes quísticos rodeados de zonas de condensación y engrosamiento perióstico en metacarpo, metatarso y falanges.
DIAGNOSTICO
Los criterios diagnósticos para el Complejo Esclerosis Tuberosa (CET) fueron revisados en la Tuberous Sclerosis Complex Consensus Conference, Julio de 1998.
CET Definitiva: Dos criterios mayores o un criterio mayor más dos criterios menores.
CET Probable: Un criterio mayor más un criterio menor.
CET Posible: Un criterio mayor o dos criterios menores.
Criterios Mayores
- Angiofibromas faciales o placas en la frente
- Fibromas no traumáticos ungueales o periungueales
- Máculas hipomelanóticas (tres o más)
- Placa de piel de zapa (nevus de tejido conectivo)
- Hamartomas nodulares retinales múltiples
- Tubérculo cortical 1
- Nódulos subependimarios
- Astrocitoma de células gigantes subependimario
- Rabdomioma cardíaco, único o múltiples
- Linfangiomiomatosis 2
- Angiomiolipoma renal 2
Criterios Menores
- Piqueteado múltiple del esmalte dental distribuido al azar
- Pólipos hamartomatosos rectales
- Quistes óseos
- Líneas de migración radial de la sustancia blanca cerebral 1,3
- Fibromas gingivales
- Hamartoma no renal
- Manchas acrómicas retinales
- Lesiones dérmicas en «Confetti»
- Quistes renales múltiples
1. La displasia cortical cerebral y las líneas de migración de la sustancia blanca cerebral si ocurren conjuntamente son contados como uno en vez de dos características de CET.
2. Cuando ambos linfangiomiomatosis y angiomiolipomas renales están presentes, otro criterio de esclerosis tuberosa debe estar presente antes de realizar el diagnóstico de CET.
3. Las líneas de migración de la sustancia blanca y la displasia cortical focal son vistos a menudo en individuos con CET; sin embargo, porque esas lesiones pueden ser vistas independientemente y son relativamente no específicas, son consideradas como un criterio diagnóstico menor para CET.
ESTUDIOS COMPLEMENTARIOS
— TAC y RMN craneal. Son imprescindibles para evaluar disfunción neurológica y la presencia simultánea de astrocitoma glial subependimario, muchas veces presente en el momento del diagnóstico inicial. Deberá ser reevaluado cada 1-3 años.
— Electroencefalograma. Sólo tiene utilidad manifiesta cuando el paciente ha comenzado su cuadro clínico inicial con crisis epilépticas.
— Ecografía renal. Debe realizarse en el momento del diagnóstico para evaluar la presencia de angiomiolipomas (28), tumores renales y/o riñones poliquísticos. Debe repetirse con una frecuencia que oscila entre 1 y 3 años.
— Electrocardiograma. Debe realizarse en el momento del diagnóstico, dada la probabilidad de desarrollar arritmias que presentan estos pacientes. El síndrome de Wolff-Parkinson-White es el más frecuente y suele presentarse tras el inicio del tratamiento con carbamacepina para el tratamiento de crisis epilépticas.
— Ecocardiograma. Se recomienda realizar ecocardiograma sólo en aquellos pacientes que muestren síntomas compatibles con rabdomioma cardíaco, puesto que las arritmias de mayor incidencia no se detectan con este método, sino mediante electrocardiograma (ECG) y las posibilidades de desarrollar disfunción cardiológica disminuyen con el crecimiento del paciente si en principio no ha presentado sintomatología adicional.
— Examen oftalmológico. Es obligado realizar fondo de ojo en el momento del diagnóstico de estos pacientes para examinar la aparición de maculopatía asociada que puede condicionar el pronóstico y calidad de vida de estos pacientes.
— Examen dermatológico. El examen dermatológico, también fundamental, es especialmente útil cuando las manifestaciones cutáneas son atípicas o cuando el diagnóstico de esclerosis tuberosa no es claro y deberemos incidir en el uso de la luz de Wood para el examen de lesiones hipocrómicas.
— Pruebas psicomotrices y de desarrollo neurológico. El panel de recomendaciones al inicio recomienda un screening de las características psicomotrices, neurológicas y comportamentales del paciente, el cual debe ser repetido en el caso de los niños en el momento de inicio de escolarización y en caso de detectar posibles anomalías a lo largo de su proceso educacional.
— Diagnóstico molecular. Las pruebas de diagnóstico molecular no están disponibles de manera rutinaria en la mayoría de los hospitales, pero en el momento que puedan llegar a estarlo la caracterización de los defectos génicos podría llegar a identificar un menor o mayor riesgo de desarrollo de las diferentes complicaciones. En un futuro será un método válido para determinar qué fenotipos se correlacionan con uno u otro defecto genético y de esta manera establecer un pronóstico acertado en el diagnóstico inicial.