
Angiosarcoma de alto grado con afectación cutánea y visceral en paciente con linfedema crónico congénito (Síndrome de Milroy)
Resumen.
Los angiosarcomas cutáneos sobre linfedema crónico congénito son tumores muy infrecuentes y presentan un comportamiento agresivo pese a la administración de tratamientos. En raros casos puede aparecer afectación visceral, lo que confiere a estos pacientes un pronóstico infausto.
Las opciones terapéuticas (radioterapia, quimioterapia, cirugía) ofrecen pobres resultados con tasas de supervivencia menores al 5% desde el momento del diagnóstico.
Autores:
Blanco Sánchez María Jesús 1
Galván Ruiz Saray 1
Dueñas Comino Arancha 2
Ros Sanjuan Laura 3
Martinez García-Cervantes Rocío 4.
- Adjunta Oncología Médica Servicio de Oncología Médica. Hospital Universitario de Gran Canaria Doctor Negrin. Las Palmas de Gran Canaria
- Residente 3º año Oncología Médica. Hospital Universitario de Gran Canaria Doctor Negrin. Las Palmas de Gran Canaria
- Residente 2º año Oncología Médica. Hospital Universitario de Gran Canaria Doctor Negrin. Las Palmas de Gran Canaria
- Residente 1º año Oncología Médica. Hospital Universitario de Gran Canaria Doctor Negrin. Las Palmas de Gran Canaria
Autor para correspondencia: María Jesús Blanco Sánchez.
Presentamos el caso de un varón de 27 años con angiosarcoma sobre linfedema crónico (enfermedad de Milroy) y afectación visceral con mala evolución a pesar de la quimioterapia administrada.
CASO CLÍNICO
Introducción:
Los angiosarcomas son tumores vasculares poco frecuentes que constituyen menos del 1% de todos los sarcomas, con predilección por la piel y partes blandas de extremidades inferiores, aunque pueden afectar a otras localizaciones 1. Se han descrito varios factores relacionados con su aparición: linfedema crónico (congénito o adquirido), radioterapia previa (especialmente en cáncer de mama), fístulas arteriovenosas, materiales extraños, injertos y enfermedades como neurofibromatosis. 2
Los angiosarcomas secundarios a linfedema crónico congénito aparecen en el 90% de los casos en pacientes con cáncer de mama tras mastectomía (síndrome de Stewart-Treves) 3,4, pero hay un mínimo porcentaje que se dan en personas con linfedema primario o congénito (enfermedad de Milroy).
Las estrategias terapéuticas actuales se basan en cirugía y radioterapia adyuvante o paliativa para control de síntomas. En cuanto al tratamiento sistémico destacar que la mayor parte de la evidencia científica proviene de series retrospectivas, lo cual hace complicado el manejo terapéutico de esta neoplasia.
El pronóstico vital en presencia de afectación visceral es infausto, con una mortalidad cercana al 100% al año.
Se presenta el caso de un varón de 27 años con angiosarcoma cutáneo sobre linfedema crónico congénito y afectación visceral con evolución fatal pese a tratamiento recibido.
Historia clínica:
Paciente varón de 27 años, con antecedentes de linfedema congénito en miembro inferior derecho (MID) desde el nacimiento en seguimiento por rehabilitación que abandonó a los 22 años. Hermano menor afecto de la misma patología.
Ingresa por dolor abdominal de dos semanas de evolución en hipocondrio derecho, destacando a la exploración física dolor a la palpación con ascitis no a tensión, linfedema crónico en miembro inferior derecho (MID) ya conocido con empastamiento a nivel inguinal y lesiones violáceas en cara medial (Imagen 1). A nivel genital se aprecia importante edema testicular con lesiones múltiples escrotales. (Imagen 2)
Se realiza linfogammagrafía con ausencia de migración en miembro inferior derecho (MID). Migración a través del sistema superficial en miembro miembro inferior izquierdo (MII).
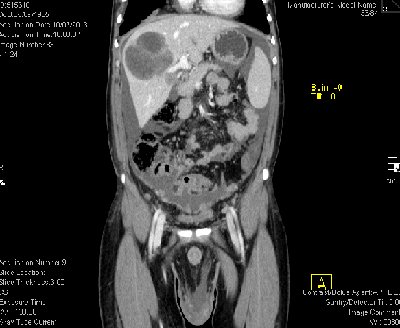
En TAC torax-abdomen-pelvis: Carcinomatosis peritoneal asociado a múltiples implantes tumorales extraperitoneales a nivel de vejiga urinaria, vesículas seminales, bolsa escrotal, pared abdominal. Derrame pleural maligno derecho con implante tumorales pleurales bilaterales. Metástasis hepáticas (imagen) y pulmonares (dudosa a nivel de la cola pancreática y del seno renal derecho). Adenopatías inguinales, retroperitoneales y mediastínicas.
– PAAF metástasis hepática: Inmunotinción positiva para vimentina y CD31 y negativa para queratinas (cóctel, AE1-AE3), PLAP, CD30, CD34, S100, HMB45, CD117 y DESMINA. Los hallazgos orientan hacia neoplasia vascular (angiosarcoma).
– Biopsia piel MID y escroto: Compatible con angiosarcoma.
El paciente inicia tratamiento con quimioterapia esquema paclitaxel dosis 80mg/m2 semanal el 23/7/13 con mala evolución, falleciendo el 7/9/13.
Juicio clínico:
Angiosarcoma cutáneo sobre linfedema crónico congénito y afectación visceral masiva asociada.
Discusión
Los angiosarcomas sobre linfedema crónico son más frecuentes en pacientes tras mastectomía por cáncer de mama (síndrome de Stewart-Treves) siendo menos habitual la asociación de esta neoplasia con linfedema crónico congénito 5,6. El linfedema congénito o enfermedad de Milroy es un trastorno autosómico dominante, que se presenta desde el nacimiento y se acompaña de historia familiar.
Su incidencia es de 1 en 6000 y se presenta con más frecuencia en el sexo femenino con una relación mujer: hombre 3:1. El origen de la enfermedad de Milroy parece encontrarse en una mutación del gen FLT4 también llamado VEGFR3 cuyo locus se encuentra en el brazo largo del cromosoma 5. Este gen es responsable de participar en los procesos de angiogénesis y linfangiogénesis y su defecto lleva a la hipoplasia o aplasia de vasos linfáticos, principalmente de miembros inferiores. Una complicación poco frecuente pero importante de linfedema crónico es el desarrollo de tumores malignos cutáneos, siendo los más frecuentes angiosarcomas.
En cuanto a su patogenia se postula que el fallo en el sistema linfático produce una alteración del sistema inmune al bloquear las células inmunocompetentes y estimular el desarrollo de una nueva red linfática y hemática. La región afectada se vuelve inmunológicamente vulnerable con mayor predisposición a desarrollar tumores vasculares dado que existe un estímulo angiogénico continuo. 7
En los angiosarcomas el tratamiento estándar es cirugía radical más radioterapia adyuvante. El papel de la quimioterapia no está claro fundamentalmente porque los estudios publicados consideran los sarcomas de partes blandas como una entidad homogénea, sin dividir por histologías, y se basan en el uso de doxorrubicina asociado o no a ifosfamida. Teniendo en cuenta que la combinación de fármacos no ha demostrado aumento de supervivencia global y sí mayor toxicidad, en la actualidad se considera la doxorrubicina en monoterapia como tratamiento de primera línea. 8 En los últimos años se están investigando fármacos más dirigidos contra determinados subtipos histológicos (imatinib en dermatofibrosarcoma protuberans, gemcitabina en leiomiosarcoma, trabectedina en liposarcoma de células redondas).
El paclitaxel a dosis semanal ha demostrado en series retrospectivas actividad en angiosarcomas, incluyéndose en las guías internacionales como alternativa a la doxorrubicina 8,9.
Un estudio fase II publicado en 2012 demuestra que paclitaxel semanal vs doxorrubicina en primera línea de angiosarcomas metastáticos presenta eficacia similar 10. En nuestro caso, el tratamiento sistémico con paclitaxel semanal no fue efectivo, falleciendo el paciente al mes y medio de inicio de la quimioterapia.
En la actualidad se están estudiando nuevos fármacos contra esta patología, fundamentalmente antiangiogénicos (bevacizumab, sorafenib, sunitinib) 11 teniendo en cuenta que la patogenia de esta enfermedad parece residir en una activación continua del sistema angiogénico. Otras dianas en investigación son la sobreexpresión de c-kit y mutación de PI3KCA.
En conclusión, los angiosarcomas cutáneos sobre linfedema crónico congénito con afectación visceral son muy infrecuentes, existiendo poca evidencia científica acerca de su manejo terapéutico. Es preciso un mayor conocimiento de las vías moleculares alteradas en este tipo de tumores para poder contar con terapias dirigidas que consigan mejorar el pronóstico, así como someter a estos pacientes a un seguimiento estricto que nos permita un diagnóstico precoz
Linfedema MID con lesión violácea.

TAC body
Bibliografía
- T. Devita, S. A. Rosenberg, and T. S. Lawrence, Cancer: Principles and Practice of Oncology, Lippincott Williams & Wilkins, Philadelphia, Pa, USA, 8th edition, 2008.
- J. Fayette, E. Martin, S. Piperno-Neumann et al., “Angiosarcomas, a heterogeneous group of sarcomas with specific behavior depending on primary site: a retrospective study of 161 cases,” Annals of Oncology, vol. 18, no. 12, pp. 2030–2036, 2007.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema; a report of six cases in elephantiasis chirurgica. Cancer. 1948 May;1(1):64-81.
- Karlsson, E. Holmberg, A. Samuelsson, K.-A. Johansson, and A. Wallgren, “Soft tissue sarcoma after treatment for breast cancer—a Swedish population-based study,” European Journal of Cancer, vol. 34, no. 13, pp. 2068–2075, 1998.
- Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, McTigue MA, et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nature genetics. 2000 Jun;25(2):153-9.
- Wananukul S, Jittitaworn S. Primary congenital lymphedema involving all limbs and genitalia. Journal of the Medical Association of Thailand = Chotmaihet thangphaet. 2005 Dec;88(12):1958-61.
- Komorowski AL, Wysocki WM, Mitus J. Angiosarcoma in a chronically lymphedematous leg: an unusual presentation of Stewart-Treves syndrome. Southern medical journal. 2003 Aug;96(8):807-8.
- Penel, A. Italiano, I. Ray-coquard et al., “Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve the outcome,” Annals of Oncology, vol. 23, no. 2, pp. 517–523, 2012
- Penel, B. N. Bui, J.-O. Bay et al., “Phase II trial of weekly paclitaxel for unresectable angiosarcoma: the ANGIOTAX study,” Journal of Clinical Oncology, vol. 26, no. 32, pp. 5269–5274, 2008
- Italiano, A. Cioffi, N. Penel et al., “Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas,” Cancer, vol. 118, no. 13, pp. 3330–3336, 2012.
- L. Ray-Coquard, J. Domont, E. Tresch-Bruneel et al., “Paclitaxel given once per week with or without bevacizumab in patients with advanced angiosarcoma: a randomized phase II trial,” Journal of Clinical Oncology, vol. 33, no. 25, pp. 2797–2802, 2015