
Presentación de un caso clínico. Talasemia mayor.
RESUMEN:
Se presenta un caso clínico de una mujer de 22 años, aquejada de una talasemia mayor presentando los siguientes síntomas: anemia, síntomas de Insuficiencia cardiaca. Los pacientes que tienen talasemia grave necesitan frecuentes transfusiones de sangre y puede que no vivan mucho tiempo.
Durante el primer año o dos primeros años de vida, pueden estar pálidos, irritables, tener poco apetito y padecer muchas infecciones. Uno de los problemas principales es la acumulación de hierro en el corazón y otros órganos, provocando insuficiencia cardiaca en algunos pacientes en los años de adolescencia o a principios de la década de los veinte. Analizaremos este caso y revisaremos su clínica y tratamiento.
Presentación de un caso clínico. Talasemia mayor.
Jiménez Cuadra, E, José María González Villalón, (Centro Salud Antequera).
Palabras clave: talasemia mayor, medicina, hematología.
SUMMARY:
We report a case of a 22 year old woman, suffering from thalassemia major presenting the following symptoms: anemia, symptoms of heart failure. Patients with thalassemia major need frequent blood transfusions and may not survive long. During the first year or first two years of life, can be pale, fussy, have a poor appetite and have many infections. One of the main problems is the accumulation of iron in the heart and other organs, causing heart failure in some patients in the teenage years or early twenties. We will discuss this case and review, his clinic and treatment.
KEY WORDS: thalasemia major, medicine, hematology.
INTRODUCCIÓN:
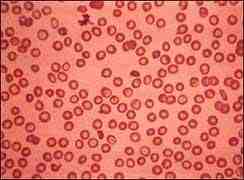
Talasemia mayor
La talasemia (1) es un trastorno hereditario que afecta a la producción de hemoglobina normal, incluye muchas formas diferentes de anemia. La gravedad y el tipo de anemia dependen del número de genes que estén afectados. La Betatalasemia es causada por mutaciones en la cadena beta de la molécula de hemoglobina. Existe un gen para la cadena beta en cada cromosoma número 11, con un total de dos genes. La forma en que estos genes están alterados determina el tipo específico de betatalasemia en una persona. Hay varios tipos:
– Beta talasemia grave (Anemia de Cooley): ambos genes de la cadena beta tienen deleciones, causando el tipo más grave de talasemia. Los pacientes que tienen talasemia grave necesitan frecuentes transfusiones de sangre y puede que no vivan mucho tiempo.
Uno de los principales problemas es la acumulación de Hierro en el corazón y otros órganos, provocando insuficiencia cardiaca en algunos pacientes en los años de adolescencia, o a principios de la década de los veinte.
– Beta talasemia leve: un gen beta, tiene una deleción provocando anemia, se divide en talasemia mínima intermedia.
HISTORIA CLÍNICA: ANAMNESIS: Se presenta un caso clínico de 22 años, presentando como clínica: esplenomegalia, hepatomegalia variable y alteraciones óseas, en cepillo y cara, implantación anómala de los dientes.
Evolución desde hace varios años. Es importante diagnosticar a tiempo las talasemias (2) para hacer un adecuado consejo genético y prevenir la aparición de casos graves de talasemia.
SÍNTOMAS: Presenta:
– esplenomegalia
– hepatomegalia variable
– fatiga
– dificultad respiratoria
Anemia (3)
DIAGNÓSTICO: Se sospechó ante la presencia de una anemia hemolítica crónica severa con microcitosis e hipocromía, y aumento de reticulocitos, pero no tanto como el que correspondería por grado de anemia (4). Por un examen físico se puede revelar la presencia de un bazo inflamado. Se tomó una muestra de sangre y se envió al laboratorio para su análisis, los glóbulos rojos aparecían pequeños y de forma anormal al examinarlos bajo un microscopio. Un hemograma mostró anemia. Un examen llamado electroforesis hemoglobínica, mostró la presencia de una forma anormal de hemoglobina. Se diagnosticó de talasemia mayor o anemia de Cooley.
TRATAMIENTO: Es fundamentalmente paliativo, (5) con la práctica de transfusiones, conviene dar quelantes de Hierro, para evitar la sobrecarga férrica, esplenectomía si compresión o hiperesplenismo. Fármacos antidrepanocíticos, que inducen la síntesis de cadenas y con aumento de la Hemoglobina F: hidroxiurea, butirato, 5-azacitidina. Los Hematíes con gran cantidad de hemoglobina (Hb) F, tienen una vida mayor y la anemia mejora. El paciente evolucionó y se mantuvo estable, mejorando con éste tratamiento.
DISCUSIÓN: CONCEPTO:
Es un trastorno sanguíneo (6) sanguíneo que se transmite de padres a hijos, en el cual el cuerpo produce una cantidad anormal de hemoglobina, la proteína en los glóbulos rojos que transporta el oxígeno.
Éste trastorno ocasiona destrucción excesiva de los glóbulos rojos, lo cual lleve a que se presente anemia. La talasemia incluye muchas formas diferentes de anemia. La gravedad y el tipo de anemia dependen del número de genes que está afectados. (7). La beta talasemia es causada por mutaciones en la cadena beta, de la molécula de la hemoglobina. Existe un gen para la cadena beta en cada cromosoma número 11, con un total de 2 genes. La forma en que estos genes, están alterados determina el tipo específico de beta talasemia en una persona.
ETIOLOGÍA: La hemoglobina se compone de 2 proteínas, la globina alfa y la globina beta. La talasemia ocurres cuando hay un defecto en un gen que ayuda a controlar la producción de una de éstas proteínas. La talasemia alfa ocurres, cuando un gen o los genes relacionados con la proteína globina (8) faltan o han cambiado. La talasemia beta ocurre cuando defectos genéticos similares afectan la producción de la proteína globina beta.
DIAGNÓSTICO Y TRATAMIENTO:
El diagnóstico de talasemia mayor beta se hace fácilmente durante la niñez, por la presencia de anemia profunda acompañada de los signos característicos de eritropoyesis ineficaz masiva: hepatoesplenomegalia, microcitosis profunda,